Identifying genetic mechanisms regulating phenotypic variability of litter size in pigs
Project funded by National Science Center (Poland) under the grant agreement 2016/23/D/NZ9/00029
Duration: 10.08.2017-9.02.2023
Summary
Research project objectives
The overall objective of the project is to increase understanding of genetic mechanisms regulating phenotypic variability of litter size in pigs by searching for genes buffering environmental factors. It has been observed that not only the mean of traits is under genetic control, but also variation around that mean, which is called the “phenotypic variability”. Genes that can buffer hard to predict (e.g., diseases) or avoid (e.g., changes in temperature) environmental factors are highly desirable, as those genes control the variation of a trait and maintain it on expected level. Those genes can also give the indication why some animals perform on the same level despite changes in the environment, when others do not.
The following detailed objectives of the project are described:
- To revise model for litter size and its phenotypic variability in order to understand the phenotypic and genetic background of variability in litter size within a sow in different parities.
- To verify previously found and detect new associations between the phenotypic variability of litter size and genotyped SNPs in pig genome.
- To identify additional genetic variants (insertions, deletions, duplications) in significant regions across population regulating phenotypic variability in litter size in pigs.
Research project methodology
Objective 1 will be studied using the traditional animal model with the application of the polynomials to fit the parity curve and Reaction Norm model which is widely used to study the mean phenotypic response to the genotype by environment interaction. This will allow applying the function of Reaction Norm to litter size (performance) in different parities of sows (environments). The results of this step will be used as basis for revised model for analysis of phenotypic variability of litter size – the Double Hierarchical Generalized Linear Model (DHGLM). With this model the differences between the animals can be studied by analyzing the residual variance of the trait and estimating its genetic variance components.
Objective 2 will be performed with application of the results from objective 1, as the DHGLM is needed to estimate the phenotypic variability of the litter size. Next the genome-wide association study (GWAS) will be performed using animals genotyped with high-density 660k SNPchip applying a multi-SNP approach using Bayesian Variable Selection method to detect new and confirm previously found regions associated with phenotypic variability of the litter size. Objective 3 will use the results of objective 2 to select animals and four genomic regions to be sequenced. The raw sequence reads will be aligned to the reference genome and subsequently scanned for variants compared to the reference genome using the pipelines available at Wageningen University. Sequenced animals will also be genotyped with the high density 660k SNP panel to ensure a better imputation of sequence variants to all animals genotyped in this study. This will also improve haplotype determination of sequenced animals. The significant SNPs in the selected regions will be detected using FreeBayes v.1.0.2 or GATK. Next the detected variants will be filtered using VCFtools v.0.1.14 and SAMtools bcftools v.1.3.
Expected impact of the research project on the development of science, civilization and society
Litter size in pigs is a complex multi-factorial trait. It is affected by many physiological aspects of the animal’s condition and its genetics. Until now only one study of Principal Investigator reported SNPs controlling the variability of this trait in pigs, but further research is needed to confirm causative mutations.
Revising the statistical model applied so far to estimate the phenotypic variability of litter size will help to account for the curve of parity effect on reproductive performance of the sow. Addressing the background of variation between parities of the sow will enable to fully understand the genetic basis of litter size variability, as it will allow to account for variability between the sows and within the production lifetime of one animal. This project is the first one to propose such approach to study variability in sow performance. The statistical methodology developed in a course of this project and knowledge could be applied to other animal species.
Increasing our understanding of genetic mechanisms regulating litter size and its phenotypic variability in pigs will facilitate knowledge on genetic architecture of those traits. Furthermore, detecting and confirming presence of genes associated with buffering environmental factors that are hard to avoid or predict will help us understand why some individuals are more susceptible to changes in the environment than others. This project is the first study aiming to detect and confirm presence of those genes in pigs. Such findings could be very important for the livestock production of the future, which needs to fulfill requirements of different production systems.
Main results
“Parity curve”
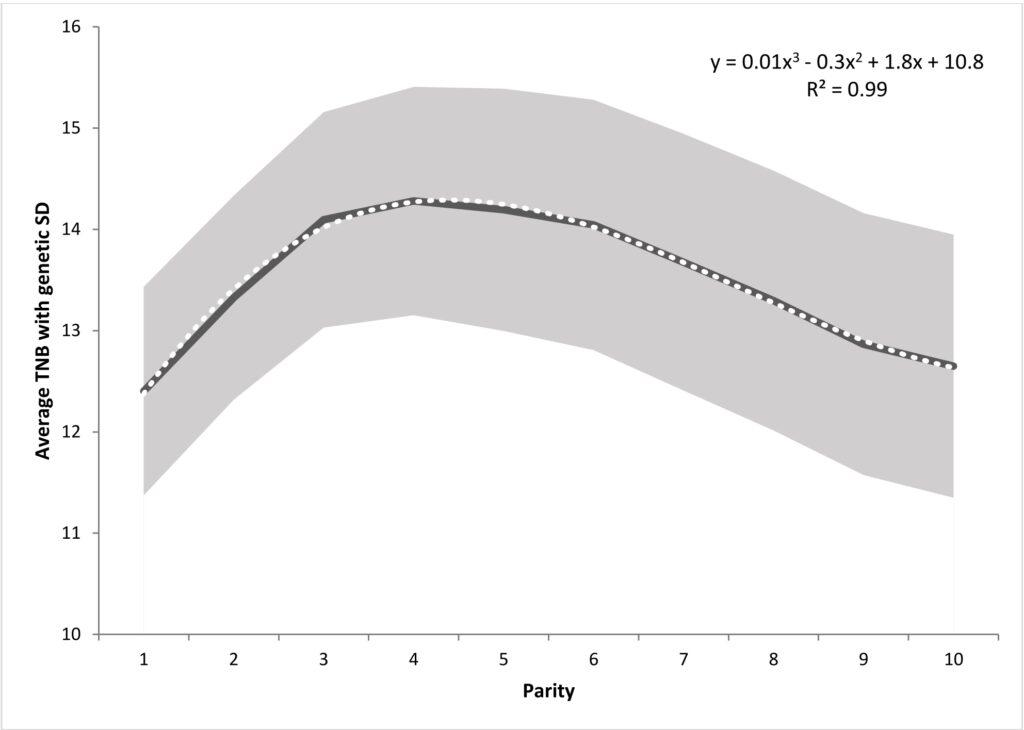
Litter size variability in successive pregnancies is non-linear. In the case of primiparous sows, litters tend to be the least numerous, after the first pregnancy, reproductive performance gradually increases, peaking between the third and fifth litters, before starting to decline again. For this reason, the name „parity curve” was adopted for this relationship. In the classic animal model, successive pregnancies are considered as one of the fixed effects. In this way, differences between the average values of the number of sow piglets due to their age are corrected. Such analysis is aimed at fitting the averaged reproductive performance curve of sows in successive pregnancies, rather than understanding the underlying differences between litter sizes between sows. Therefore, in Sell-Kubiak et al. (2019) a model was used that allowed to include a „parity curve” in the analysis of litter size. Analogous to the milk yield analyzes of the cows, a third-order Legendre polynomial random regression model was used for the additive genetic effect and the sustainable environmental effect to fit the sow’s “parity curve”.
This model was compared to a very simplified model with a „parity curve” as a fixed effect only. In the publication of Sell-Kubiak et al. (2019) indicated that when comparing the Akaike criterion for both models, it was determined that the random regression model with polynomials for random effects was better suited to litter size than the classic animal model. Studies have shown that both genetic variance and heritability increase with successive pregnancies, starting with the second pregnancy. Estimated genetic correlations indicate that the first pregnancy differs the most from pregnancies 7-10, while it is most similar to pregnancies 2 and 3. In general, the values of genetic and phenotypic correlations indicated a strong relationship between subsequent pregnancies. Moreover, the obtained genetic parameters „proved” the existence of significant genetic variability around the „parity curve”, which would suggest the possibility of further selection for this trait. Subsequent analyzes using the previously obtained variance components from the random regression model in the simulations of the breeding index showed that changing the shape of the „parity curve” is practically impossible or carries unfavorable consequences.
First, selection to increase litter size in the first pregnancy would increase the size of all subsequent pregnancies, but would not affect the shape of the “parity curve”. Second, selecting for more piglets in the first gestation while selecting for smaller litters in the fifth gestation, while reducing the differences between successive pregnancies, would also reduce the average litter size in the overall population. The above studies indicated that there is genetic variability around the “parity curve” of sows. However, it is not possible to assume in the breeding program that selection in the direction of changing the shape of this curve will make it possible to reduce the variation in litter size between successive pregnancies. This is due to the fact that there are very strong genetic and phenotypic relationships between individual pregnancies, which are very difficult to eliminate even when the litter size of one of the parities is selected.
"Best model to estimate litter size variability"
"Real background of second litter syndrome"
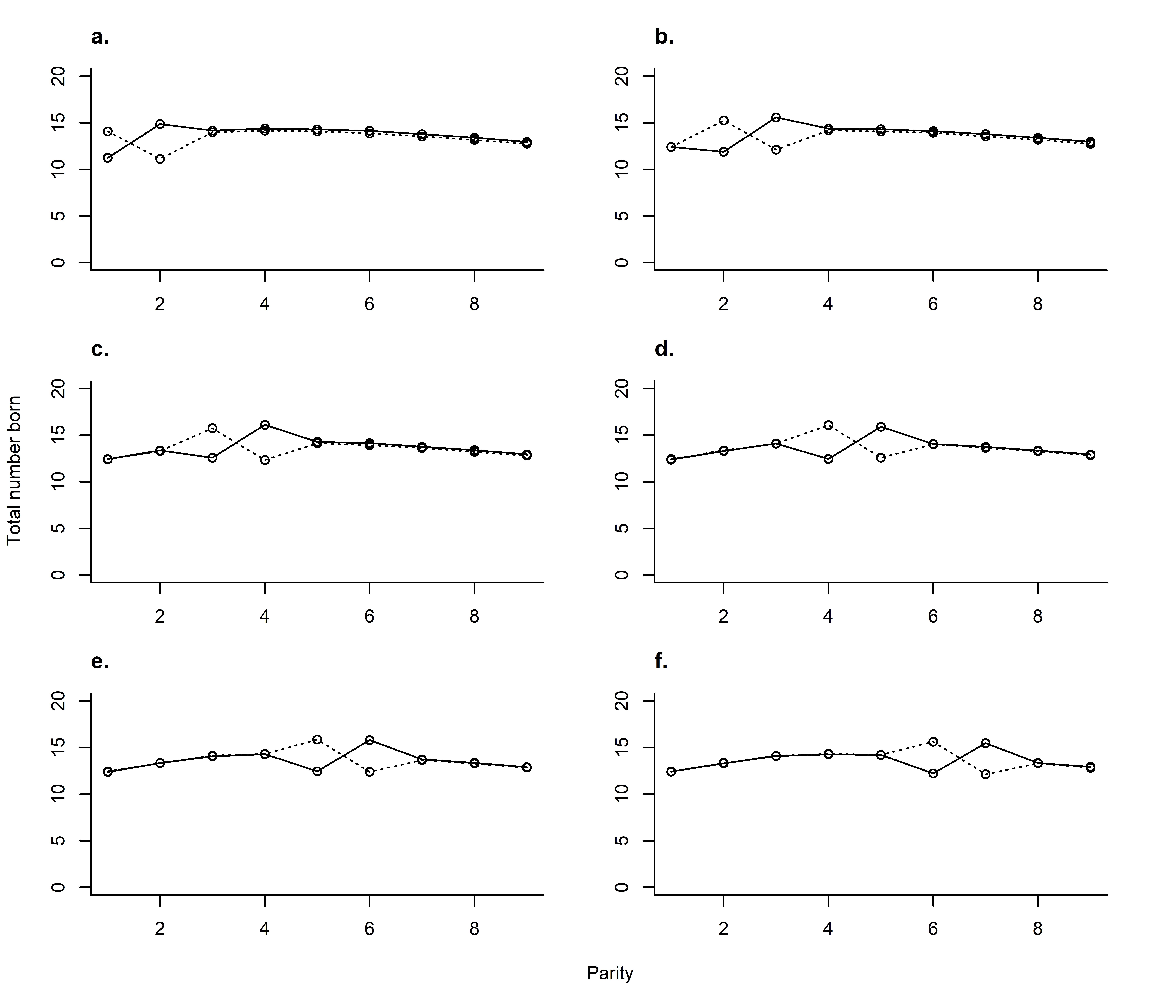
Since litter size is an extremely important characteristic in pig production, any deviation from the expected mean value characteristic for a given pregnancy, i.e. the “consecutive litter curve”, is of concern to farmers. One of these problems is the so-called second litter syndrome (SLS), which describes a situation where a sow in the second litter produces fewer piglets than in the first litter. The literature indicates a range of 20 to 60% of such cases in pig herds of various breeds. Considering the significant scale of the problem and its negative impact on the number of piglets obtained from young sows in a litter, research into the causes of this phenomenon has been conducted for many years. However, this work has primarily focused on factors related to the sow’s environment and condition, and never on the potential genetic basis.
Research presented by Sell-Kubiak et al. (2021) showed that the only significant risk factors for SLS were farm-related factors (lactation length, farm specificity, and time of year of second farrowing). This means that the breeder has an influence on the higher frequency of SLS, and it is not a physiological problem of the animal itself. Although the research allowed to estimate the genetic component of two traits defining SLS (biSLS – binary trait, Range – continuous trait), and therefore heritability was also obtained for them (for biSLS it was 0.05, and for Range 0.03) , this is not evidence of a genetic basis for second litter syndrome. Estimated heritability is related to genetic variation around the „litter size curve” and should not be interpreted as the genetic basis of SLS, but as another means of comparing litter-to-sow variation. Further research based on simulations showed that the average expected frequency of SLS in all parities was 0.49 (±0.05), while the observed frequency was 0.46 (±0.04). What’s more, we can expect a smaller number of piglets in the next litter compared to the previous one with the same probability, because it results from the characteristics of the normal distribution.
Assuming, according to empirical observations, that the size of the first litter is on average smaller than the size of the second litter, the chance of SLS occurrence is less than 50%. This is in line with my observations in the Great White sow population where SLS occurred at a rate of 30%. In addition, a comparison of the incidence of SLS on the 67 farms from which the sows used in my analyzes came, showed that only two of them had, on average, more piglets in the first litter than in the second. This again pointed to issues related to herd management rather than reproductive predisposition of the sows. The main conclusion of this work is to show that individual cases of SLS are caused by statistical properties of litter size, while at the farm level, the occurrence of SLS is probably due to management errors. Therefore, SLS should not be considered as a „syndrome” that should be specifically eliminated from herds. On the other hand, the breeder should monitor the cases of the second litter being smaller in relation to the first one and react with a change in management if their number increases alarmingly.
"Consequences of further selection towards increased litter size"
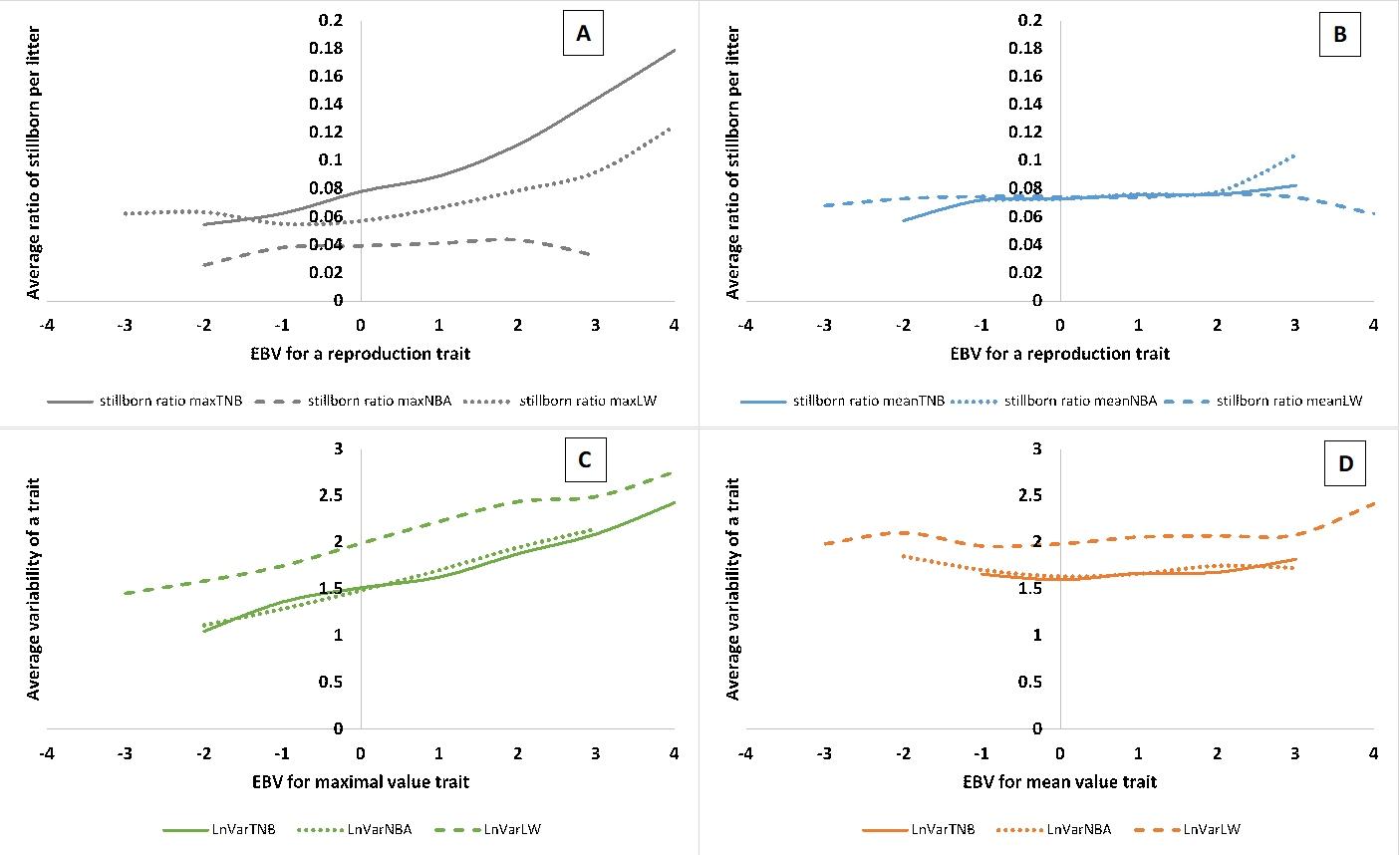
Pig farming has reached a stage where litter size, birth weight and the balance between the two require a balanced approach to better care for the well-being of sows and piglets. What’s more, we also observe that with the increasing number of litters, pig farmers have considerable difficulties with rearing piglets. Therefore, in Sell-Kubiak (2021), the characteristics related to the maximum litter size and weight were examined. The maximum value of the examined traits had a significantly higher heritability than the traits analyzed classically (i.e. taking into account every available observation) or the average of these values, e.g. numbers, which would suggest a faster response to selection. However, the maximum value traits carried a higher risk of stillborn piglets (although not for the maximum number of live-born piglets) and increased variability in all reproductive traits tested. Therefore, the use of maximum values in breeding programs should be carefully considered and should be considered as an assessment of the sow’s potential, rather than a parameter included in the breeding index.
Therefore, in the further part of the work, the maximum values of the features were not taken into account. Genetic correlations between the average values of reproductive traits and the corresponding variability traits ranged from 0.66 to 0.74, while the remaining correlations ranged from 0.33 to 0.99. Such high genetic correlations meant that in the simulations of selection indices, even with selection aimed solely at unifying reproductive traits, a very limited change should be expected due to the strong genetic and phenotypic relationship between the traits. Although from a genetic point of view selection aimed at increasing both litter size and birth weight is still possible, intensification of this selection by selecting „maximum values of traits” is not advisable. Moreover, for reasons of animal welfare and herd management, it would be much better to select a balance between the average values of reproductive traits and the diversity of litter size and weight, which can only be achieved through a very well-developed breeding program.
"Selection of the statistical method to analyze litter size variability"
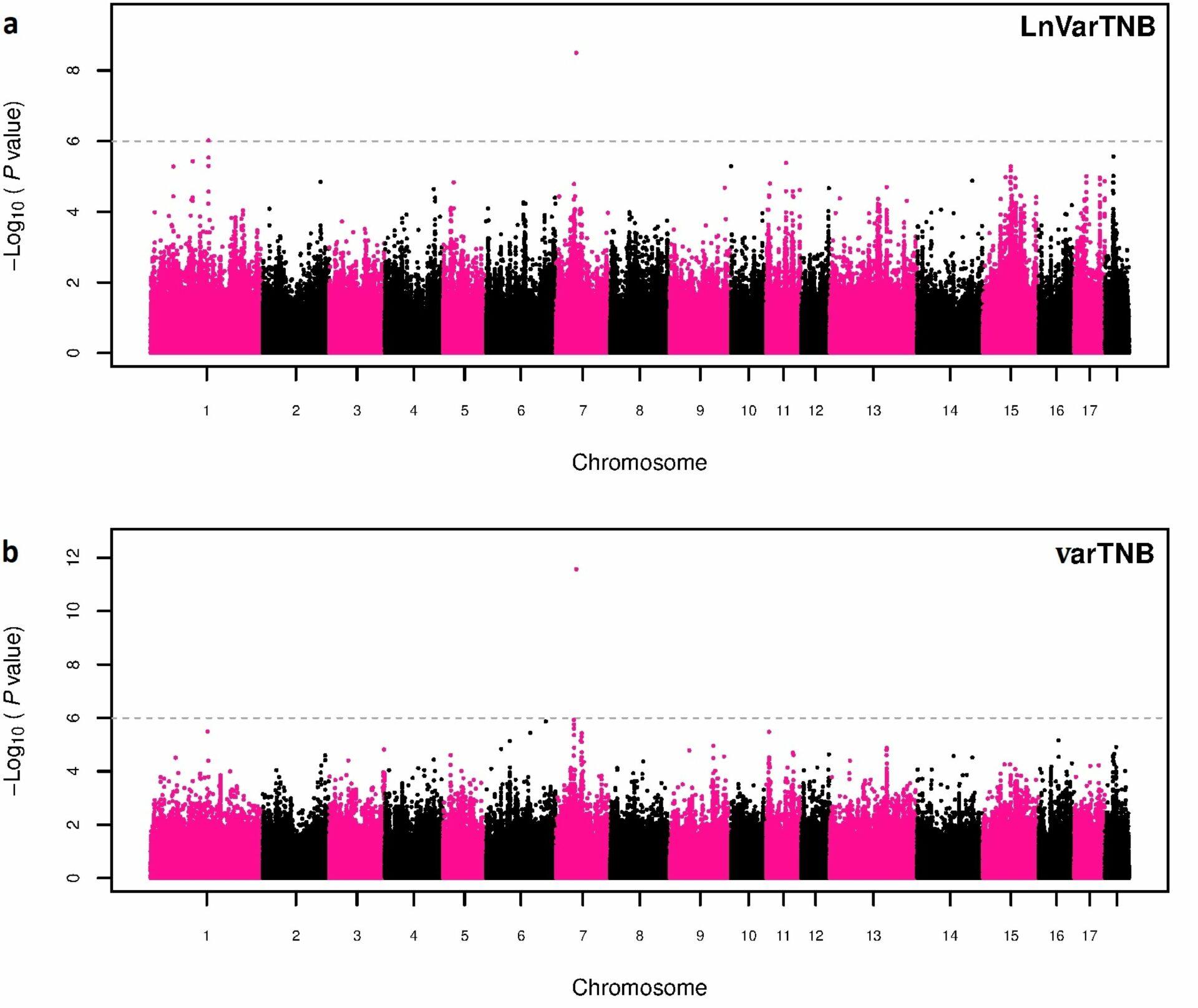
Based on phenotypic data used in Sell-Kubiak et al. (2022a) showed that a simpler method of determining the variance phenotype (natural logarithm of the residual variance – derived from a classical animal model – of all sow observations, LnVar) is less accurate than the more complicated Double Hierarchical Generalized Linear model Mixed model, DHGLM). This was confirmed by: lower correlation between breeding values obtained in 2021 and those from 2015 estimated on a smaller number of animals (0.73 for LnVar and 0.86 for DHGLM); lower accuracy of obtained breeding values; poorer accuracy in estimating breeding values for unobserved individuals. Interestingly, despite the differences between the two methods, a genome-wide association study identified the same region in the porcine genome as the most significant for both traits (Sell-Kubiak et al., 2022a). It was one SNP on chromosome 7 located in an intron important for regulatory reasons for the porcine genome in the so-called Genomic Evolutionary Rate Profiling (GERP).
Potential candidate genes located within +/- 50,000 base pairs from the most important SNP are: ADGRF1 responsible for the neurological development of the brain and ADGRF5 responsible for the proper functioning of the respiratory system and the formation of blood vessels. The use of much larger data resources in this study than in the 2015 paper confirmed that the most important region in controlling litter size variation is chromosome 7. The linkage between the SNPs identified in 2015 and now is 0.11. In view of the above, it can be concluded that both in research and breeding programs, DHGLM should be used to analyze the genetic diversity of the phenotype of traits, as it is a much more accurate method than the simpler method of determining these phenotypes. On the other hand, chromosome 7 in the pig genome remains unquestionably the most important region determining litter size variation.
"Meta-analysis of SNPs related to reproductive traits"
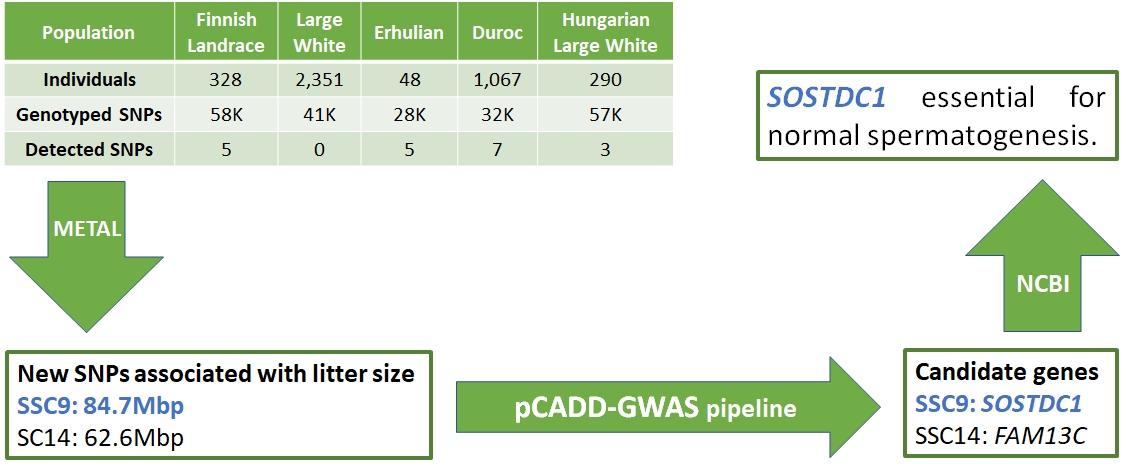
Sell-Kubiak et al. (2022b) used data from previous studies of single nucleotide polymorphisms associated with litter size to conduct a comprehensive meta-analysis. Based on published genome-wide association studies (GWAS), we know that there are at least 2,000 SNPs associated with pig litter size traits. The aim of the meta-analysis was to collect and integrate previously published associations between SNPs and five reproductive traits: total birth number (TNB), number of live births (NBA), number of stillborns (SB), birth weight of litter (LWT) and corpus luteum (CLN). ), to verify their common genetic background and perform a meta-analysis of the total number of births (TNB) GWAS from five GWAS studies for the TNB trait. Only 21 of the 233 candidate genes described in previous publications were identified in more than one population or for more than one trait. Based on these observations, the most interesting candidate gene was PRKD1, which is associated with SB and TNB traits.
Based on a subsequent analysis using GO terms, it was shown that PRKD1 is also involved in angiogenesis. Nevertheless, the results of this part of the study showed how highly polygenic pig reproductive traits are and how few genes are pleiotropic. A meta-analysis of GWAS studies identified two single nucleotide polymorphisms (SNPs) in the porcine genome that had not previously been reported in other studies. One SNP was located on chromosome 14 in the intron of the FAM13C gene. The second SNP was located on chromosome 9 within the intron of the AGMO gene. Further functional analysis identified SOSTDC1 as a strong candidate gene, associated with reduced fertility in male rats. This is a very important result because litter traits are assumed to be associated with females, not males. Therefore, it is possible that boars have a much greater influence on the number of piglets born than we believe.
"Genomic and phenotypic characterization of litter size variability in Landrace pigs"
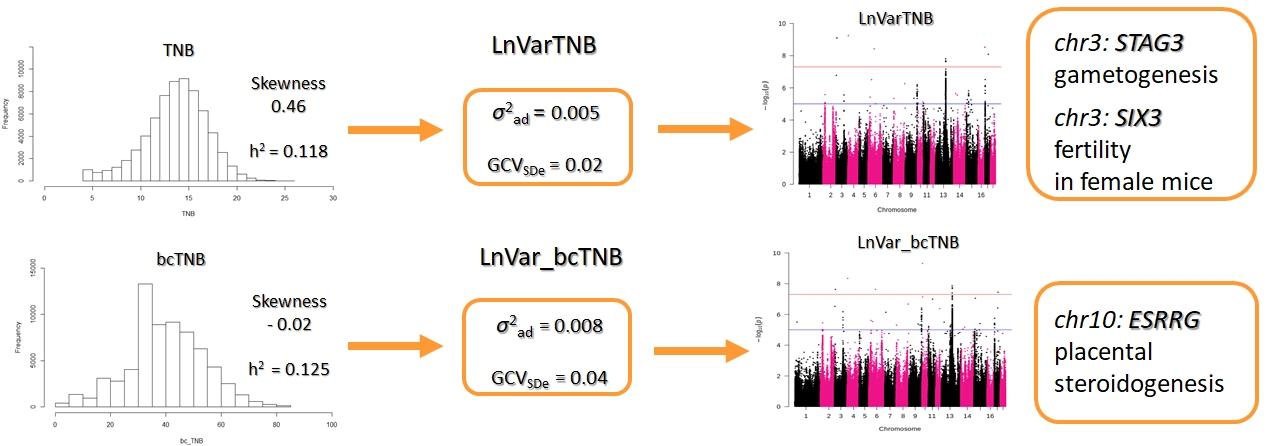
Subsequent studies aimed to assess the phenotypic and genomic background of litter size variability obtained from data before and after Box-Cox transformation (Cieleń et al., in press). Since the empirical distribution of the analyzed features showed significant skewness, it was decided to perform the Box-Cox transformation and obtain bcTNB. The phenotypic variation was then estimated as LnVar for both TNB and bcTNB. The heritability for TNB was 0.118 compared to 0.125 for bcTNB. In contrast, the heritability for LnVar_TNB was 0.0025 compared to 0.0037 for LnVar_bcTNB. The difference between TNB before and after transformation was also indicated in the genetic correlation between mean litter size and its variability changed from 0.38 (LnVar_TNB) to 0.63 (LnVar_bcTNB). Based on the GWAS, 14 SNPs for LnVar_TNB and eight for LnVar_bcTNB were detected, with two of them located at the most promising candidate genes.
The first of them is a gene located on chromosome 3 – STAG3, which plays an important role in gametogenesis. The second gene on chromosome 10 is ESRRG, which affects the development of the placenta. Additional analysis of the GWAS results revealed another promising candidate gene located on chromosome 13, MFN1, involved in embryonic development. The results of this study showed a significant change in the components of variance for the variability when the Box-Cox transformation was applied to data showing skewness. Moreover, data transformation changed the phenotype enough that only part of the SNPs overlapped the two variability traits. Our research shows that it is necessary to perform a Box-Cox transformation on skewed data in order to properly estimate the phenotypic and genomic characteristics of the variation of the trait.
"Genomic and phenotypic characterization of litter size variability in Landrace pigs"
Publications and conference proceedings
Publications
Sell-Kubiak, E., J. Dobrzanski, M. F. L. Derks, M. S. Lopes and T. Szwaczkowski. 2022. Meta-Analysis of SNPs Determining Litter Traits in Pigs. Genes, 10(13):1730. doi: 10.3390/genes13101730.
Sell-Kubiak, E., E. F. Knol, M. S. Lopes. 2022. Evaluation of phenotypic and genomic background of trait variability based on large-scale litter size data of Large White pigs. Genetics Selection Evolution, 54(1). doi: 10.1186/s12711-021-00692-5.
Sell-Kubiak, E., E. F. Knol, H. A. Mulder, M. Pszczoła. 2021. Unraveling the actual background of second litter syndrome in pigs based on Large White data. Animal, 15(2):100033. doi: 10.1016/j.animal.2020.100033.
Sell-Kubiak, E. 2021. Selection for litter size and litter birthweight in Large White pigs: maximum, mean and variability of reproduction traits. Animal, 15(10):100352. doi: 10.1016/j.animal.2021.100352.
Dobrzański, J., H. A. Mulder, E. F. Knol, T. Szwaczkowski, and E. Sell-Kubiak. 2020. Estimation of litter size variability phenotypes in Large White sows. Journal of Animal Breeding and Genetics, 137:559–570. doi: 10.1111/jbg.12465.
Sell-Kubiak, E., E. F. Knol, H. A. Mulder. 2019. Selecting for changes in average “parity curve” pattern of litter size in Large White pigs. Journal of Animal Breeding and Genetics, 136(2): 134-148. doi: 10.1111/jbg.12372.
Conferences proceedings
Cieleń, G., E.F. Knol, M.S. Lopes, E. Sell-Kubiak. 2022. To Box-Cox or not to Box-Cox: phenotypic and genomic evaluation of litter size variability in Landrace pigs. WCGALP, Rotterdam, the Netherlands.
Derks, M., A. Boshove, B. Harlizius, E. Sell-Kubiak, M.S. Lopes, E. Grindflek, E. Knol, M.A.M. Groenen, A.B. Gjuvsland. 2022. A pan-genome of commercial pig breeds. WCGALP, Rotterdam, the Netherlands.
Sell-Kubiak, E., E. F. Knol, H. A. Mulder, M. Pszczoła. 2021. Omówienie podłoża syndromu drugiego miotu. LXXXV Zjazd Polskiego Towarzystwa Zootechnicznego, Olsztyn, Poland.
Sell-Kubiak, E., E. F. Knol, M. S. Lopes. 2021. High-density genome-wide association for variability of litter size in pigs. Book of Abstracts of the EAAP, Davos, Switzerland.
Derks, M. F. L., B. Harlizius, M. van Son, E. Sell-Kubiak, M. S. Lopes, E. Grindflek, E. F. Knol, A. B. Gjuvsland. 2021. A pangenome of commercial pig breeds. Proceedings of the 38th International Society for Animal Genetics Virtual Conference (ISAG).
Sell-Kubiak, E., E. F. Knol. 2020. “Maximum vs. Optimum”: litter size and birthweight in pigs. Book of Abstracts of the EAAP Virtual Meeting.
Dobrzański, J., H. A. Mulder, E. F. Knol, T. Szwaczkowski, E. Sell-Kubiak. 2019. Different models for estimation of litter size variability phenotypes. Book of Abstracts of the EAAP, Ghent, Belgium.
Pszczoła, M., E. F. Knol, H. A. Mulder, E. Sell-Kubiak. 2019. Verifying the existence of second litter syndrome in pigs. Book of Abstracts of the EAAP, Ghent, Belgium.
Dobrzański, J., H. A. Mulder, E. F. Knol, T. Szwaczkowski, E. Sell-Kubiak. 2018. Applying Different Models to Estimate Litter Size Variability in Large White Sows. Book of abstracts XXVIII Genetic Days, České Budějovice, Czech Republic.
Sell-Kubiak, E., E. F. Knol, H. A. Mulder. 2018. The genetic background of second litter syndrome in pigs. Book of Abstracts of the EAAP, Dubrovnik, Croatia.
Sell-Kubiak, E. 2018. Genetic background of litter size variability in pigs. 1st Conference Animal Genetic and Genomics, Olsztyn, Poland. (invited speaker)
Scientific titles
Habilitation – Ewa Sell-Kubiak, 23.09.2022
“Identyfikacja genetycznych mechanizmów kontrolujących zmienność fenotypową liczebność miotu świń”
MSC – Gabriela Cieleń, 30.06.2022
“Genome-wide association study for litter size variability in Landrace pigs”
Non-scientific publications and interviews
Sell-Kubiak, E. 2021. Możliwe ograniczenia płynące z selekcji w kierunku maksymalizacji liczebności i masy miotu. Trzoda chlewna 11:22-25.
Sell-Kubiak, E. 2021. Czy syndrom drugiego miotu faktycznie istnieje? Trzoda chlewna 2:18-19
Wywiady dla Polskiej Agencji Prasowej:
- Naukowcy wesprą hodowców świń – opublikowany 24.05.2018 r. scienceinpoland.pap.pl
- Badacze doskonalą hodowlę zwierząt gospodarskich – opublikowany 24.08.2016 r. naukawpolsce.pap.pl